CHARGE
: Atomic charges from difference density
Author: Nick Spadaccini,
Computer Science Department, University of Western Australia,
Nedlands, 6907 WA, Australia
CHARGE calculates a charge associated with an
atom from the difference density. The difference density
(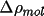 ) is partitioned
according to the Hirshfeld method.
) is partitioned
according to the Hirshfeld method.
The Hirshfeld method apportions the electron density
among the atoms by the appropriate weighting. The weights
are related by the atomic contribution to the promolecular
density,
|
(4.1) |
![\[
w_{A}(\Vector{r}) = \frac{\rho_{\rm{atm}}^A(\Vector{r})} { \rho_{\rm{pro}}(\Vector{r})}
\]](equations/4.12.1-eq-1.gif) |
The fragment of the deformation density
apportioned to atom A is,
|
(4.2) |
![\[
\Delta\rho_{\rm{frag}}^A(\Vector{r}) = w_{A}(\Vector{r})
\Delta\rho_{\rm{mol}}(\Vector{r})
\]](equations/4.12.1-eq-2.gif) |
The net atomic charge,
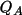 , is derived from the
integration of the difference density fragment,
, is derived from the
integration of the difference density fragment,
|
(4.3) |
![\[
Q_{A} = \int_{\rm{input}} {\Delta\rho_{\rm{frag}}^{A}(\Vector{r})d\Vector{v} }
\]](equations/4.12.1-eq-3.gif) |
An alternative scheme is based on the
atomic contributions to the total promolecular potential
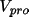 defined as the sum of
the electronic and nuclear contributions.
defined as the sum of
the electronic and nuclear contributions.
Density And Potential Profiles
The promolecular density or potential is the sum of
the atomic densities or potentials. These latter values are
derived from the Clementi and Roetti atomic wavefunctions.
Associated with each atom type are the parameters
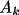 ,
,
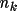 and
and
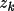 for k=1,...,m such that the density is,
for k=1,...,m such that the density is,
|
(4.4) |
![\[
\rho_{\rm{atm}}(\Vector{r}) =\sum_{k=1}^{m}{ A_k r^{n_k}e^{-z_k r} }
\]](equations/4.12.2-eq-4.gif) |
and the potential is
|
(4.5) |
![\[
V_{\rm{atm}}(\Vector{r}) =4\pi \sum_{k=1}^{m}{ A_k \left(
\frac{\gamma(n_k+3,z_k r) }{r z_{k}^{n_k+3} } +
\frac{(n+1)! }{ z_{k}^{n_k+2} } -
\frac{\gamma(n_k+2,z_k r) }{ z_{k}^{n_k+2} } \right)
+ \frac{Z_A}{r} }
\]](equations/4.12.2-eq-5.gif) |
The last term is the nuclear contribution and
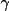 (n,x) is the
Incomplete Gamma Function.
(n,x) is the
Incomplete Gamma Function.
The density profiles (e/
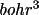 ) and potential
profiles (e/bohr) are stored at 44 discrete values of r
(bohrs) for the points,
) and potential
profiles (e/bohr) are stored at 44 discrete values of r
(bohrs) for the points,
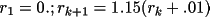 ; 0. ≤ r ≤
31.2
; 0. ≤ r ≤
31.2
The divisions are chosen so that the density of
points is greatest in the region of steepest gradient. The
density or potential value at any general point is linearly
interpolated from the profile.
Density, Distances And Errors
The difference density (
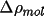 ) must be input from the
FOURR file
) must be input from the
FOURR file
map. CHARGE partitions
this density into atomic contributions and integrates over
the input region to obtain a charge. If the input map is
the asymmetric unit the values obtained are total atomic
charges for atoms at general positions or fractions
governed by the site symmetry for atoms at special
positions. The user must determine the fraction of an atom
present in the input map and calculate the total charge
accordingly.
The user may specify the effective range of atom
contributions in two ways. The
border
option in the program initiation line
determines the region beyond the input map for which atom
contributions are included. The default value of 6Å
implies that any atom within this distance of the input map
edges is included. Also the distance beyond which an atom
contribution is zero may be set by the
contact
option.
Estimates of
 (Q) are determined
for spherical regions of various radii following the method
of Davis and Maslen. The estimates are derived from the
values of
(Q) are determined
for spherical regions of various radii following the method
of Davis and Maslen. The estimates are derived from the
values of
 (F), the errors in
the structure factor amplitudes used to derive the
difference density. For each atom the program outputs a
value of
(F), the errors in
the structure factor amplitudes used to derive the
difference density. For each atom the program outputs a
value of
 (Q) for a radius
set within the program. The absolute radii are generated
from relative radii, derived from the density profiles and
stored in the database. These values are rescaled so that
the total volume of the atoms in the cell equals the cell
volume. The radii used are listed.
(Q) for a radius
set within the program. The absolute radii are generated
from relative radii, derived from the density profiles and
stored in the database. These values are rescaled so that
the total volume of the atoms in the cell equals the cell
volume. The radii used are listed.
Since values of atomic radii are not unique the
variation of
 (Q) with r is also
output so that the user may determine
(Q) with r is also
output so that the user may determine
 (Q) at an
alternative radius if desired. The scheme assumes a
centrosymmetric structure so that phase errors are not
included
.
(Q) at an
alternative radius if desired. The scheme assumes a
centrosymmetric structure so that phase errors are not
included
.
-
Reads
lrcell:
and symmetry data from the input archive
bdf
-
Reads difference density from a
mapfile
-
Reads profile data base from the file
designated by the macro
profile:
The weights here are determined from the
promolecular density. The contributions of atoms up to
6Å beyond the input map edges are included in the
calculation. However, an atom will not contribute to the
density if the point is greater than 6Å away.
Here the weights are determined from the
promolecular potential. Atoms up to 4Å from the
input map edges are included in the calculation. The
effective range of a atom is set to 4.5Å.
-
Clementi, E and Roetti, C. 1974.
Atomic Data and Nuclear Data
Tables
14, 177-478.
-
Davis, C.L. and Maslen, E.N. 1978.
Acta Cryst.
A34, 743-746.
-
Hirshfeld, F. 1977.
Israel Journal of Chemistry
16, 198-201.





