|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| atrad | atomic radii by atom type |
| addpek | add peak site to atom list from
pekfile |
| bond | calculate a specific bond distance |
| angle | calculate a specific bond angle |
| dihed | calculate a specific dihedral angle |
| nviron | calculate distances to a specific atom site |
| site | enter temporary atom sites needed for geometry |
| BONDLA | Option | Code | Arg | Def | |
| bond distances |
bond
|
list all bond distances | |||
|
no bond distances | ||||
| bond angles |
angl
|
list all bond angles | |||
|
no angles | ||||
| contact distances |
ncon
|
no contact distances | |||
|
list all contact distances | ||||
| dihedral angles |
ndih
|
no dihedral angles | |||
|
list all dihedral angles | ||||
| bond check |
|
for Acta Cryst CIF bond checking | |||
| sort bond & contact distances |
nsor
|
do not sort distances | |||
|
sort distances | ||||
| optimally connect input sites |
clus
|
recluster input sites | |||
|
use input sites | ||||
| use full symmetry to searches |
symm
|
apply full symmetry | |||
|
no symmetry | ||||
| do not output geom data to bdf |
|
output tobdf | |||
use brackets in
output file |
|
no brackets | |||
| number of cells searched |
|
|
1for
1 unit cell |
3
|
|
2for
27 cells |
|||||
3for
125 cells |
| atrad | 1 | atom-type symbol to which the radii apply(max 8 chars) | |
| 2 | maximum radius to be used in identifying contacts |
1.25
|
|
| 3 | maximum radius to be used in identifying bonds |
0.9
|
|
| 4 | minimum radius to be used in identifying bonds |
0.015
|
| addpek | 1 | atom-type symbol assigned to selected peak sites (max 8 chars) | |
| 2 | number of peaks assigned to atom-type |
1
|
|
| 3 | maximum radius to be used in identifying contacts |
1.25
|
|
| 4 | maximum radius to be used in identifying bonds |
0.9
|
|
| 5 | minimum radius to be used in identifying bonds |
0.015
|
|
| 6 | thermal displacement parameter assigned to output site |
0.035
|
| bond | 1-2 | atom labels of sites 1 and 2 (max 24 chars) | |
| 3-4 | numbers [a]of symops applied to sites 1 and 2 |
1
|
|
| 5-6 | codes of cell translations applied to sites 1 and 2 |
555
|
|
[a] Symmetry matrix number as entered into STARTX. | |||
| angle | 1-3 | atom labels of sites 1, 2 and 3 (max 24 chars) | |
| 4-6 | numbers [a]of symops applied to sites 1, 2 and 3 |
1
|
|
| 7-9 | codes of cell translations applied to sites 1, 2 and 3 |
555
|
|
[a] Symmetry matrix number as entered into STARTX. | |||
| dihed | 1-4 | atom labels of sites 1, 2, 3 and 4 (max 24 chars) | |
| 5-8 | numbers [a]of symops applied to sites 1, 2, 3 and 4 |
1
|
|
| 9-12 | codes of cell translations applied to sites 1, 2, 3 and 4 |
555
|
|
[a] Symmetry matrix number as entered into STARTX. | |||
| nviron | 1 | atom label of central site (max 24 chars) | |
| 2 | number [a]of symop applied to sites |
1
|
|
| 3 | code of cell translations applied to site |
555
|
|
| 4 | radius of sphere to be searched for other sites |
3
|
|
[a] Symmetry matrix number as entered into STARTX. | |||
| site | 1 | atom label of site [a](max 24 chars) | |
| 2-4 | fractional coordinates x, y, z | ||
| 5 | U |
0.035
|
|
| 6 | population |
1
|
|
| 7-9 |
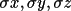
|
0.0
|
|
[a] If label matches site on the bdf, these coordinates will be substituted for the calculation. Otherwise a new site will be added to the atom list. This site will not be added to the bdf. | |||

 Overview News
Overview News  Example GraphicsHistory
Example GraphicsHistory Documentation
Documentation ![]() Xtal Users Manual
Xtal Users Manual![]()
![]() Preface
Preface![]()
![]() Primer
Primer![]()
![]() Program controls
Program controls![]()
![]()
![]() System
System![]()
![]()
![]()
ABSORB
![]()
![]()
![]()
ADDATM
![]()
![]()
![]()
ADDPAT
![]()
![]()
![]()
ADDREF
![]()
![]()
![]()
ATABLE
![]()
![]()
![]()
BAYEST
![]()
![]()
![]()
BONDAT
![]()
![]()
![]()

BONDLA

![]()
![]()
![]()
BUNYIP
![]()
![]()
![]()
CBAZA
![]()
![]()
![]()
CHARGE
![]()
![]()
![]()
CIFIO
![]()
![]()
![]()
CIFENT
![]()
![]()
![]()
CONTRS
![]()
![]()
![]()
CREDUC
![]()
![]()
![]()
CRILSQ
![]()
![]()
![]()
CRISP
![]()
![]()
![]()
CRYLSQ
![]()
![]()
![]()
DIFDAT
![]()
![]()
![]()
DYNAMO
![]()
![]()
![]()
EXPAND
![]()
![]()
![]()
FC
![]()
![]()
![]()
FOURR
![]()
![]()
![]()
GENEV
![]()
![]()
![]()
GENSIN
![]()
![]()
![]()
GENTAN
![]()
![]()
![]()
GIP
![]()
![]()
![]()
LATCON
![]()
![]()
![]()
LISTFC
![]()
![]()
![]()
LSABS
![]()
![]()
![]()
LSLS
![]()
![]()
![]()
LSQPL
![]()
![]()
![]()
LSRES
![]()
![]()
![]()
MAPLST
![]()
![]()
![]()
MAPXCH
![]()
![]()
![]()
MODEL
![]()
![]()
![]()
MODHKL
![]()
![]()
![]()
NEWCEL
![]()
![]()
![]()
NEWMAN
![]()
![]()
![]()
ORTEP
![]()
![]()
![]()
OUTSRC
![]()
![]()
![]()
PARTN
![]()
![]()
![]()
PATSEE
![]()
![]()
![]()
PEKPIK
![]()
![]()
![]()
PIG
![]()
![]()
![]()
PLOTX
![]()
![]()
![]()
POWGEN
![]()
![]()
![]()
PREABS
![]()
![]()
![]()
PREPUB
![]()
![]()
![]()
PREVUE
![]()
![]()
![]()
REFCAL
![]()
![]()
![]()
REFM90
![]()
![]()
![]()
REGFE
![]()
![]()
![]()
REGWT
![]()
![]()
![]()
RFOURR
![]()
![]()
![]()
RIGBOD
![]()
![]()
![]()
RMAP
![]()
![]()
![]()
RSCAN
![]()
![]()
![]()
SCATOM
![]()
![]()
![]()
SHAPE
![]()
![]()
![]()
SHELIN
![]()
![]()
![]()
SIMPEL
![]()
![]()
![]()
SLANT
![]()
![]()
![]()
SORTRF
![]()
![]()
![]()
STARTX
![]()
![]()
![]()
SURFIN
![]()
![]()
![]()
VUBDF
![]()
![]()
![]()
XTINCT
![]()
![]() Reference Manual
Reference Manual![]()
![]() Archive
Archive![]()
![]() Symmetry
Symmetry![]() FAQ
FAQ![]() Developer Info
Developer Info![]() Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer
Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer