|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| disper | dispersion factor |
| scale | scale factor |
| extinc | extinction parameters |
| maxhkl | reflection limits |
| xabs | absolute-structure parameter |
| select | select subset of atoms from the bdf |
| dptype | change atomic displacement parameter type |
| group | group or identical molecule refinement |
| atomgr [a] | atoms per group |
| restr [b] | establish conditions for positional parameters |
| constr [b] | constrain one parameter to be a function of otherparameters |
| noref [b] | shut off refinement of specified parameters |
| block | matrix blocking line |
|
[a] Must be preceded by a group line [b] Order specific lines. restr lines precede constr which precede noref lines. | |
| CRYLSQ | Option | Code | Arg | Def | |
| number of refine cycles |
|
|
1
|
||
| refinement coefficient |
fr
|
F | |||
|
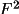
|
||||
|
intensity | ||||
|
F(heavy atom) | ||||
| derivative matrices |
bd
|
block-diagonal | |||
|
full-matrix | ||||
|
specified in block lines | ||||
| reflection weight |
ws
|
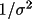
|
|||
|
unit weights | ||||
|
|
bdf wt with id=190
n
|
0
|
||
| scale factor(s) |
us
|
refine together | |||
|
refine separately | ||||
| dispersion |
|
apply but do not refine | |||
|
apply and refine | ||||
| extinction |
|
apply but do not refine | |||
|
apply and refine | ||||
| abs-struct. param |
|
apply but do not refine | |||
|
apply and refine | ||||
| thermal displacement type |
mx
|
mixed iso and aniso (as in bdf) | |||
|
isotropic | ||||
|
anisotropic | ||||
|
overall | ||||
| population |
|
yes | |||
| dataset number |
|
|
data set number |
bdf or
1
|
|
| reflection skip |
|
|
use every
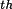 reflection reflection |
||
| fixed atoms used |
|
apply values from prior run | |||
|
apply & store on output bdf | ||||
| type hkl as rcode2 [a] |
|
|
only if Y <
 Y Y |
||
| use in matrix |
|
|
only if w
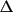 Y < Y <
|
||
| rcode2 use in matrix |
|
only if
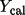 > >
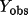 |
|||
| shift damping factor |
|
|
for
|
0.8
|
|
for
|
1.0
|
||||
| non positive-def. U |
tr
|
make pos. def. and continue | |||
|
tell user and continue | ||||
|
terminate calculation | ||||
| termination criterion |
|
stop if no convergence of R | |||
| list reflections |
|
last cycle | no | ||
|
every cycle | ||||
| list poor reflections |
|
|
if w
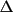 Y > Y >
|
||
atom lines to
pchfile |
|
last cycle | no | ||
|
every cycle | no | |||
| list correl. matrix |
|
|
if > q after the last cycle | ||
| list deriv. matrices |
|
after last cycle only | |||
|
after every cycle | ||||
| save correl. matrix |
|
output to file
cmx
|
|||
| old atomic parameters |
|
output old params to the bdf | |||
[a] Applies for the current run only and does not affect the bdf. | |||||
| disper | 1 | atom-type name | |
| 2 | real part of dispersion scattering factor | ||
| 3 | imaginary part of dispersion scattering factor | ||
| 4-6 | as in 1-3, or use separate disper lines |
| extinc | 1 | extinction correction method | |||
| Zachariasen |
zach
|
||||
| Becker-Coppens, type I |
|
||||
| Becker-Coppens, type II |
|
||||
| Becker-Coppens, type I and II |
|
||||
| 2 | extinction type | ||||
| isotropic |
is
|
||||
| anisotropic (not implemented) |
|
||||
| 3 | mosaic distribution | ||||
| Gaussian distribution |
gaus
|
||||
| Lorentzian distribution |
|
||||
| 4 | isotropic extinction parameter(
zachor
typ1) |
(g) |
0
|
||
| 5 | isotropic extinction parameter(
typ2or
gen) |
(
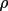 ) ) |
0
|
||
| 6 | mean value of T in mm | Xray |
0.3
|
||
| neutron |
1.5
|
| maxhkl | 1-3 | max |h|, |k|, |l| applied in refinement process |
999
|
| 4 | min value of sin
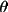 / /
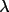 applied applied |
0
|
|
| 5 | max value of sin
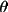 / /
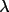 applied applied |
10
|
| select | 1- | atom label or atom label string [a] | |
[a] An atom label string is two atom labels separated by a slash '/'. The first label identifies the first atom , and the second label the last atom in a sequence of atoms. | |||
| dptype | 1 | atom type symbol | |||
| 2 | thermal displacement type | ||||
| overall U |
|
||||
| isotropic U |
|
||||
anisotropic
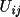 |
|
||||
| 3-4 | as in 1, 2, or use separate dptype lines |
| group | 1 | group refinement number of molecule |
0
|
||
| 2-4 | x, y, z fractional coordinate of centre of gravity, if fixed | ||||
| 5 | Euler angle refinement signal | not fixed | |||
| Euler angles invariant |
0
|
||||
| symmetry element is parallel or perp. to a-axis |
|
||||
| symmetry element is parallel or perp. to b-axis |
|
||||
| symmetry element is parallel or perp. to c-axis |
|
||||
| 6 | signal for the refinement of isotropic U | ||||
| group isotropic U |
+1
|
||||
| invariant U |
|
||||
| individual isotropic U |
|
||||
| 7 | signal for population parameter refinement | no constraints | |||
| group population |
|
||||
| invariant population |
0
|
||||
| individual population |
|
| atomgr | 1 | atom label string [a] | |
[a] An atom label string is two atom labels separated by a slash '/'. The first label identifies the first atom , and the second label the last atom in a sequence of atoms. | |||
| restr [a] | 1 | restraint class | |||
| bond length |
|
(n=2 atoms) | |||
| bond angle |
|
(n=3 atoms) | |||
| dihedral angle |
|
(n=4 atoms) | |||
| 2 | atom labels of the n restrained atoms [b] | ||||
| n+2 | ideal values of bond length, angle or dihedral angle | ||||
| n+3 | standard deviation of ideal value | ||||
|
[a] restr is an order specific line and cannot be preceded by constr or noref lines. | |||||
| constr [a] | 1 | contraints equation
[b]
(see
CRYLSQ
) |
|
|
[a] constr is an order specific line and cannot be preceded by noref lines. [b] Embedded blanks are allowed. The order of the constraints is not arbitrary - reference atoms must occur higher in the atom list than subject atoms. Furthermore, for parameters belonging to the same atom, the reference parameters must occur earlier in the parameter list than the subject parameters. The parameters may be any of the following: SKF, UOV, EXT, DSP, X, Y, Z, U, U11, U22, U33, U12, U13, U23, POP, APP, and NEU. | |||
| noref | 1 | invariant parameter string [a] | |
[a] No embedded blanks are allowed. See the constr line for the list of parameters and for special atom names. | |||
| block [a] | 1 | matrix block ( same format as noref ) | |
[a] Any block lines must cover all selected parameters. When no parameter symbol is given, a new block is started. | |||

 Overview News
Overview News  Example GraphicsHistory
Example GraphicsHistory Documentation
Documentation ![]() Xtal Users Manual
Xtal Users Manual![]()
![]() Preface
Preface![]()
![]() Primer
Primer![]()
![]() Program controls
Program controls![]()
![]()
![]() System
System![]()
![]()
![]()
ABSORB
![]()
![]()
![]()
ADDATM
![]()
![]()
![]()
ADDPAT
![]()
![]()
![]()
ADDREF
![]()
![]()
![]()
ATABLE
![]()
![]()
![]()
BAYEST
![]()
![]()
![]()
BONDAT
![]()
![]()
![]()
BONDLA
![]()
![]()
![]()
BUNYIP
![]()
![]()
![]()
CBAZA
![]()
![]()
![]()
CHARGE
![]()
![]()
![]()
CIFIO
![]()
![]()
![]()
CIFENT
![]()
![]()
![]()
CONTRS
![]()
![]()
![]()
CREDUC
![]()
![]()
![]()
CRILSQ
![]()
![]()
![]()
CRISP
![]()
![]()
![]()

CRYLSQ

![]()
![]()
![]()
DIFDAT
![]()
![]()
![]()
DYNAMO
![]()
![]()
![]()
EXPAND
![]()
![]()
![]()
FC
![]()
![]()
![]()
FOURR
![]()
![]()
![]()
GENEV
![]()
![]()
![]()
GENSIN
![]()
![]()
![]()
GENTAN
![]()
![]()
![]()
GIP
![]()
![]()
![]()
LATCON
![]()
![]()
![]()
LISTFC
![]()
![]()
![]()
LSABS
![]()
![]()
![]()
LSLS
![]()
![]()
![]()
LSQPL
![]()
![]()
![]()
LSRES
![]()
![]()
![]()
MAPLST
![]()
![]()
![]()
MAPXCH
![]()
![]()
![]()
MODEL
![]()
![]()
![]()
MODHKL
![]()
![]()
![]()
NEWCEL
![]()
![]()
![]()
NEWMAN
![]()
![]()
![]()
ORTEP
![]()
![]()
![]()
OUTSRC
![]()
![]()
![]()
PARTN
![]()
![]()
![]()
PATSEE
![]()
![]()
![]()
PEKPIK
![]()
![]()
![]()
PIG
![]()
![]()
![]()
PLOTX
![]()
![]()
![]()
POWGEN
![]()
![]()
![]()
PREABS
![]()
![]()
![]()
PREPUB
![]()
![]()
![]()
PREVUE
![]()
![]()
![]()
REFCAL
![]()
![]()
![]()
REFM90
![]()
![]()
![]()
REGFE
![]()
![]()
![]()
REGWT
![]()
![]()
![]()
RFOURR
![]()
![]()
![]()
RIGBOD
![]()
![]()
![]()
RMAP
![]()
![]()
![]()
RSCAN
![]()
![]()
![]()
SCATOM
![]()
![]()
![]()
SHAPE
![]()
![]()
![]()
SHELIN
![]()
![]()
![]()
SIMPEL
![]()
![]()
![]()
SLANT
![]()
![]()
![]()
SORTRF
![]()
![]()
![]()
STARTX
![]()
![]()
![]()
SURFIN
![]()
![]()
![]()
VUBDF
![]()
![]()
![]()
XTINCT
![]()
![]() Reference Manual
Reference Manual![]()
![]() Archive
Archive![]()
![]() Symmetry
Symmetry![]() FAQ
FAQ![]() Developer Info
Developer Info![]() Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer
Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer