|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| label | descriptive information |
| cell | unit cell parameters |
| cellsd | standard deviations in unit cell parameters |
| sgname [a] | space group name in explicit-origin Hall notation |
| spaceg | space group name in Hermann-Maugin notation |
| latice [a] | unit cell lattice type and centricity |
| symtry [a] | symmetry equivalent positions |
| celmol | number of molecules in the cell |
| celcon | contents of unit cell atomic species information |
| formfx [b] | scattering factor information, tabular |
| formgn [b] | scattering factor information, expansion coefficients |
| scale [c] |
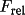 scale factors
for data sets scale factors
for data sets |
| maxhkl [c] | maximum
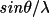 , h, k, and l for
datasets , h, k, and l for
datasets |
| datdef [c] | definition of crystal data set (enantiomorph, etc.) |
| exper [c] | experimental conditions for data sets |
| crystal | crystal colour and dimensions |
| orient [c] | crystal orientation for data sets |
|
[a] Symmetry information must be entered by using either a sgname line or a latice line and/or one or more symtry lines. [b] CELCON lines must precede these lines. For a given atom type, both formfx and a formgn lines may not be used. However, a formfx line for atom type 1 and a formgn line for atom type 2 may be used. If neither is entered, stored expansion coefficients will be used by default for atoms specified on the CELCON lines (Cromer and Mann, 1968). [c] These lines, if entered, must follow the CELCON lines. | |
| STARTX | Option | Code | Arg | Def | |
| bdf creation |
apr
|
create an a priori output archive bdf | |||
|
output updated from input bdf | ||||
| stored form factors |
|
|
sin
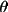 / /
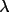 interpolation int. interpolation int. |
0.02
|
|
|
|
sin
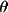 / /
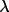 maximum maximum |
2.0
|
||
| radiation type |
xray
|
X-rays | |||
|
neutrons | ||||
|
electrons | ||||
|
no symmetry checking at all |
| label | 1 | character information added to the logical
record
lrlabl:
of the bdf |
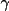



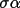
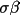
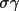
| sgname | 1- | space group in Hall notation [a] | |
| spaceg | 1- | space group in Hermann-Maugin notation [a] | |
[a] Separate elements of the notation must be separated by space or underscore characters e.g. P 1 21 2 not P1211. Note also that because colons (:) denote specific settings they cannot be used as XTAL comment delimiters on spaceg lines. | |||
| latice [a] | 1 | centricity code | |||
| centrosymmetric |
|
||||
| noncentrosymmetric |
|
||||
| 2 | Bravais lattice code | ||||
| primitive |
|
||||
| end-centred |
,
,
|
||||
| body-centred |
|
||||
| rhombohedral |
|
||||
| face-centred |
|
||||
| 3 | no symmetry checks [b] |
|
|||
|
[a] If this line is entered then the equivalent positions described by the centricity and Bravais lattice codes must not be input as symtry lines. This line must NOT be entered if an sgname line is used. [b] no symmetry checks will be applied to input symtry lines and the sgname code will not be automatically generated and stored. This option is invoked if special symmetry is to be entered. | |||||
| symtry [a] | 1- | equivalent positions entered as algebraic strings | |
[a] Symmetry equivalent positions are entered as listed in the International Tables, Vol I or A. If the latice line is entered then the equivalent positions described by the centricity and Bravais lattice codes must not be input as symtry lines. The definition of an equivalent position is the x, y, and z operations for each coordinate separated by commas. Only one equivalent position may be defined on a symtry line. This line must NOT be entered if an sgname line is used. Xtal comments (preceded by a colon :) are not permitted. | |||
| celcon [a] | 1 | atom type symbol (max 8 characters) | |
| 2 | number of atoms of this type in the unit cell | ||
| 3 | atomic weight | based on field 1 | |
| 4 | atomic number | based on field 1 | |
| 5 | number of electrons in atom type | based on field 1 | |
| 6 | atomic bond radius (in Å) | based on field 1 | |
| 7 | atomic contact radius (in Å) | based on field 1 | |
| 8 | real dispersion factor f' [b] | based on field 1 | |
| 9 | imaginary dispersion factor f" [b] | based on field 1 | |
| 10 | neutron scattering factor (CAUTION [c]) [d] | based on field 1 | |
|
[a] One celcon line must be entered for each atom type expected in the structure. If neither formgn nor formfx lines are used, scattering factors will be generated using information on celcon lines and tables from Cromer and Mann (1968). [b] The dispersion factors are automatically
entered only for MoK and CuK
[c] If the neutron scattering factor is entered, ALL subsequent calculations will require that neutron data be processed. Do not enter unless this is the case. [d] If the command STARTX neut is invoked then the neutron scattering length is automatically added to the archive. | |||
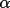
| formfx [a] | 1 | atom type symbol (max 8 characters) | |
| 2 |
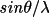 for scattering
factor for scattering
factor |
||
| 3 | scattering factor value | ||
| 4 | comment about source of scattering factor (surround with '=') | ||
[a] Not more than 200
formfx lines
are allowed per atom type. Lines must be in
order of increasing
| |||
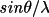
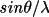
| formgn [a] | 1 | atom type symbol (max 8 characters) | |
| 2-10 | Cromer-Mann coefficients a1, b1, a2, b2 , a3, b3, a4, b4, c | ||
[a] One formgn line is used per atom type. | |||
| scale | 1 |
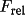 scale scale |
|
| 2 | scale group number |
1
|
|
| 3 | data set number |
1
|
|
| 4-6 | same as fields 1-3 for next scale; and so on |
| maxhkl | 1-3 | max |h|, |k|, |l| to be stored on the bdf | |
| 4-5 | min and max
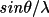 to be stored on the
bdf to be stored on the
bdf |
||
| 6 | data set number |
1
|
| exper | 1 | data set number |
1
|
| 2 | weighted mean wavelength (in Å) |
0.71069
(X rays)
|
|
1.0(neutrons)
|
|||
0.01(electrons)
|
|||
| 3 | wavelength line 1 | ||
| 4 | wavelength line 2 | ||
| 5 | wavelength line 3 | ||
| 6 | relative weight line 1 | ||
| 7 | relative weight line 2 | ||
| 8 | relative weight line 3 | ||
| 9 | measured density of crystal (mg/
 ) ) |
||
| 10 | linear absorption coefficient | ||
| 11 | temperature of measurement (in Celsius) | ||
| 12 | sorting order for hkl as packed integer e.g.
213is
k-slow, l-fast |
||
| 13 | pressure of measurement (in GPa) | ||
| 14 | radiation type (1=neutron 2=electron 3=MoKa 4=CuKa 5=AgKa 6=synch) | ||
| crystal | 1 | crystal colour (20 characters max) | |
| 2 | crystal shape (20 characters max) | ||
| 3 | Maximum crystal dimension (mm) | ||
| 4 | Median crystal dimension (mm) | ||
| 5 | Minimum crystal dimension (mm) | ||

 Overview News
Overview News  Example GraphicsHistory
Example GraphicsHistory Documentation
Documentation ![]() Xtal Users Manual
Xtal Users Manual![]()
![]() Preface
Preface![]()
![]() Primer
Primer![]()
![]() Program controls
Program controls![]()
![]()
![]() System
System![]()
![]()
![]()
ABSORB
![]()
![]()
![]()
ADDATM
![]()
![]()
![]()
ADDPAT
![]()
![]()
![]()
ADDREF
![]()
![]()
![]()
ATABLE
![]()
![]()
![]()
BAYEST
![]()
![]()
![]()
BONDAT
![]()
![]()
![]()
BONDLA
![]()
![]()
![]()
BUNYIP
![]()
![]()
![]()
CBAZA
![]()
![]()
![]()
CHARGE
![]()
![]()
![]()
CIFIO
![]()
![]()
![]()
CIFENT
![]()
![]()
![]()
CONTRS
![]()
![]()
![]()
CREDUC
![]()
![]()
![]()
CRILSQ
![]()
![]()
![]()
CRISP
![]()
![]()
![]()
CRYLSQ
![]()
![]()
![]()
DIFDAT
![]()
![]()
![]()
DYNAMO
![]()
![]()
![]()
EXPAND
![]()
![]()
![]()
FC
![]()
![]()
![]()
FOURR
![]()
![]()
![]()
GENEV
![]()
![]()
![]()
GENSIN
![]()
![]()
![]()
GENTAN
![]()
![]()
![]()
GIP
![]()
![]()
![]()
LATCON
![]()
![]()
![]()
LISTFC
![]()
![]()
![]()
LSABS
![]()
![]()
![]()
LSLS
![]()
![]()
![]()
LSQPL
![]()
![]()
![]()
LSRES
![]()
![]()
![]()
MAPLST
![]()
![]()
![]()
MAPXCH
![]()
![]()
![]()
MODEL
![]()
![]()
![]()
MODHKL
![]()
![]()
![]()
NEWCEL
![]()
![]()
![]()
NEWMAN
![]()
![]()
![]()
ORTEP
![]()
![]()
![]()
OUTSRC
![]()
![]()
![]()
PARTN
![]()
![]()
![]()
PATSEE
![]()
![]()
![]()
PEKPIK
![]()
![]()
![]()
PIG
![]()
![]()
![]()
PLOTX
![]()
![]()
![]()
POWGEN
![]()
![]()
![]()
PREABS
![]()
![]()
![]()
PREPUB
![]()
![]()
![]()
PREVUE
![]()
![]()
![]()
REFCAL
![]()
![]()
![]()
REFM90
![]()
![]()
![]()
REGFE
![]()
![]()
![]()
REGWT
![]()
![]()
![]()
RFOURR
![]()
![]()
![]()
RIGBOD
![]()
![]()
![]()
RMAP
![]()
![]()
![]()
RSCAN
![]()
![]()
![]()
SCATOM
![]()
![]()
![]()
SHAPE
![]()
![]()
![]()
SHELIN
![]()
![]()
![]()
SIMPEL
![]()
![]()
![]()
SLANT
![]()
![]()
![]()
SORTRF
![]()
![]()
![]()

STARTX

![]()
![]()
![]()
SURFIN
![]()
![]()
![]()
VUBDF
![]()
![]()
![]()
XTINCT
![]()
![]() Reference Manual
Reference Manual![]()
![]() Archive
Archive![]()
![]() Symmetry
Symmetry![]() FAQ
FAQ![]() Developer Info
Developer Info![]() Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer
Retired Program InfoDownloadExtra SoftwareSupport ContributorsCitationsYear 2000Disclaimer