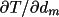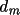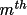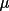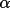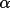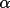|
LSABS
: Absorption corrections
Authors: Eric Blanc &
Dieter Schwarzenbach
Contact: Eric Blanc, Institut
de Cristallographie, University of Lausanne, 1015 Lausanne,
Switzerland
LSABS calculates transmission factors T,
absorption-weighted mean-path lengths T, and derivatives,
Methods of Absorption Correction
LSABS offers two methods
of integration, the analytical method and Gaussian
integration. The analytical method is more efficient than
the Gaussian method in calculating the derivatives. For
this reason, execution times for the two methods are
comparable in the case of a coarse Gaussian grid, and in
favour of the analytical method in the case of a fine
Gaussian grid.
The analytical method proposed by de Meulenaer &
Tompa (1965) describes the crystal shape by the set of its
vertices, whereas
LSABS defines it by the
set of its edges. This technique facilitates an unambiguous
identification of the vertices and minimizes rounding
errors.
Gaussian grid integrationThe implementation of the numerical integration of
Gauss-Legendre offers a choice of grids with 6, 8, 10 ,12,
14, 16, 24, or 32 intervals for each dimension. For needle-
and plate- shaped crystals the orientation of the grid may
be selected to lie along one of the principal directions.
For a needle, the z-axis of the grid is chosen along the
needle axis identified by its zone indices [
u v w ]. For a plate, the z-axis of the
grid is chosen along the plate normal identified by the
face indices (
h k l ). The calculation of the
derivatives
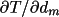 requires evaluation of
surface integrals. Consequently a surface grid must be
specified in addition to the 3-dimensional grid for the
volume integral. requires evaluation of
surface integrals. Consequently a surface grid must be
specified in addition to the 3-dimensional grid for the
volume integral.
Input and Output Data SpecificationThe M plane faces of a polyhedral crystal are defined
by their Miller indices (
h
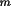 k
k
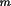 l
l
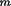 ) relative to the lattice base
a,
b,
c, and by the
perpendicular distances
d ) relative to the lattice base
a,
b,
c, and by the
perpendicular distances
d
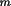 from a given point inside the crystal to each face
(m = 1, ..M). For the natural faces of a crystal, (
h from a given point inside the crystal to each face
(m = 1, ..M). For the natural faces of a crystal, (
h
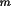 k
k
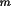 l
l
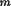 ) are integers.
d ) are integers.
d
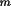 should have been carefully measured and an
estimated standard deviation of
d should have been carefully measured and an
estimated standard deviation of
d
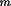 should be available. The I Bragg reflections are
identified by their Miller indices
h should be available. The I Bragg reflections are
identified by their Miller indices
h
 k
k
 l
l
 and an instrument-independent crystal-based
azimuthal angle and an instrument-independent crystal-based
azimuthal angle
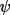
 as defined by Schwarzenbach & Flack (1989,
1992). The above data must be available on the bdf for
LSABS to function
correctly. as defined by Schwarzenbach & Flack (1989,
1992). The above data must be available on the bdf for
LSABS to function
correctly.
By default the nett intensities of the reflections
(Schwarzenbach & Flack, 1991) are not modified by
LSABS : the calculated
quantities A=1/T, <T>(i.e. Tbar) and
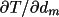 are simply stored on the
bdf. are simply stored on the
bdf.
The recommended method of placing raw diffractometer
information including crystal face indices and distance
data on the bdf is first to transform a
diffractometer-specific file into a CIF using DIFRAC
(Flack, Blanc & Schwarzenbach, 1992). This file may
then be read with
CIFENT
(same input lines as
CIFIO
) to create a bdf. This process compactly loads all
of the necessary raw experimental data onto a bdf in a
diffractometer-independent way.
The above example shows the use of
STARTX
,
REFCAL
, LSABS, and
SORTRF
to create a bdf with |
F
 | data from diffractometer data produced in CIF
format by the DIFRAC program. | data from diffractometer data produced in CIF
format by the DIFRAC program.
STARTX
calculates the direct and reciprocal cell metrics,
loads the scattering factor tables, atomic radii, and
checks the symmetry information - the
sgname line is necessary
in
STARTX
as this information is not stored with the raw
diffractometer data.
REFCAL
calculates the reflection information.
LSABS performs a Gaussian
grid integration and applies the absorption correction to
the net intensities in the bdf. As the crystal is
needle-shaped, the Gaussian grid is oriented with the
needle direction [1 2 0] parallel to the grid
z axis. A finer grid is defined along the needle axis than
in the other directions. A second pass through
REFCAL
is needed to convert the net intensities to |
F
 | corrected for absorption. Finally the
absorption-corrected data are sorted and averaged leaving a
bdf ready for structure solution and conventional |
F | corrected for absorption. Finally the
absorption-corrected data are sorted and averaged leaving a
bdf ready for structure solution and conventional |
F
 | refinement. | refinement.
A bdf with net intensity data is created.
LSABS performs an
analytical absorption correction and stores the resulting
values in the bdf. The bdf is suitable for the refinement
of the crystal shape and decay parameters.
-
Blanc, E., Schwarzenbach, D. & Flack, H.D.
(1991).
J. Appl. Cryst.
24, 1035-1041
-
Busing, W. R. & Levy, H. A. (1957).
Acta Cryst.
10, 180-182.
-
de Meulenaner, J. & Tompa, H. 1965.
Acta Cryst.
19,
1014-1018.
-
Flack, H.D., Blanc, E. & Schwarzenbach, D.
(1992)
J. Appl. Cryst.
25, 455-459.
-
Ibers, J.A. & Hamilton, W.C. (1974) Eds.
International Tables for X-rays
Crystallography Vol. IV, Birmingham,
England: Kynock Press
-
Schwarzenbach, D. & Flack, H. D. (1989).
J. Appl. Cryst.
22, 601-605.
-
Schwarzenbach, D. & Flack, H. D. (1991).
Acta Cryst.
A47, 134-137.
-
Schwarzenbach, D. & Flack, H. D. (1992).
J. Appl. Cryst.
25, 69.
|