RMAP
:
Search for translated fragment
Authors: William Furey and
Jim Stewart
Contact: Jim Stewart,
Department of Chemistry, University Maryland, College Park,
MD 20742 USA
RMAP calculates R factors or correlation
coefficients as a function of the translated position of a
molecular fragment in the unit cell. In addition to the
translated fragment there may also be a fixed fragment.
Control is provided for selecting reflections by resolution
magnitude of Fobs, or numbers. The step size and range the
unit cell to be used in the search also may be
specified.
RMAP is used to calculate maps analogous to electron
density maps where each point in the map is a function of
1/R, where R is the conventional R factor (
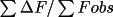 ). The use of 1/R forces the coordinate
of best agreement to be on a maximum. In addition to 1/R
maps it is possible to calculate correlation maps (referred
to as C maps), or a linear function of 1/R and the
correlation. The correlation C function is
). The use of 1/R forces the coordinate
of best agreement to be on a maximum. In addition to 1/R
maps it is possible to calculate correlation maps (referred
to as C maps), or a linear function of 1/R and the
correlation. The correlation C function is
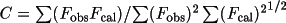 at each grid
point.
at each grid
point.
The advantage of C is the fact that it is independent
of the Frel scale factor. It is not as "sharp" a function
as 1/R, however. It also requires more storage for the
additional sums. The maps will show a maximum at the
optimum translational position of a fragment in the unit
cell. A grid, just like the grid in a regular Fourier
transform, is set up in a specified region of the unit cell
and structure factors are calculated for a selected subset
of reflections at each of the grid points. The values of
the calculated structure factors are used to calculate R or
C or a linear combination of R and C, at each grid point. A
linear combination which gives numbers on a convenient
scale has been defined as:
D = {10(1-W)C+1}{(W/R)+1}
The values of D are written to a
MAPfile in the same
format as that produced by
FOURR
.
MAPmay be used with
PEKPIK
to search for the maxima. The highest maximum is
reported in the RMAP output. The factor W is a factorial
weight. If W=1, only R contributes to D, while if W=0, only
the correlation coefficient contributes to D. When W=1 is
used, 1/3 the amount of storage is required.
It is not usually necessary, or desirable, to include
all reflections in the search procedure. Much time can be
saved if only the strong reflections, at an appropriate
level of resolution, are used in the promotion of D values.
It may be necessary to include a small number of weak
reflections to control the scale factor. Perhaps a better
method for this is to use
GENEV
with all data to establish a physically realistic
Frel scale factor before using RMAP. It is useful to start
with low resolution data and a coarse grid. The program is
set to use 0.3 Angstrom steps over the whole unit cell with
all reflections included in the calculation as default
parameters.
Initially when no positional information is available
the number of atoms in the fixed fragment will be zero and
the calculation will be made only on the translated
fragment. After a fragment has been positioned, the first
fragment may be declared as fixed and any additional
fragment atoms can be designated as translated. This
feature allows molecules to "grow" and allows use of the
program where there is more than one molecule in the
asymmetric portion of the unit cell.
In the initial pass over the bdf data the
contributions of all the atoms to the structure factor
components A and B, as a function of the symmetry
operations, are saved as separate quantities. The selected
reflections are written to a scratch file. On the second
pass the structure factors are calculated as a function of
all the grid points. This is accomplished by using the sums
of A and B at each symmetry point and the trigonometric
identities:
cos(
x +
y ) = cos
x cos
y - sin
x sin
y
sin(
x +
y ) = cos
x sin
y + sin
x cos
y
From these identities each step
y can be applied to each geometrical A
and B part of the structure factor (
cosx, sinx ) and the complete structure
factor at the step formed by summing only over the number
of symmetry operations and not over the number of atoms in
the fragment.
The input bdf must have
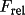 or
or
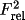 , and a set of atomic
coordinates with an overall or with individual isotropic
thermal displacement parameters. The atoms must be loaded
(with
, and a set of atomic
coordinates with an overall or with individual isotropic
thermal displacement parameters. The atoms must be loaded
(with
ADDATM
) as one or two subsets. The first subset, which may
be null, contains the atoms of a fragment which is to
remain fixed during the search. The second subset (which
must not be null) consists of the atoms to be translated
during the search.
Atom sites are listed by entering
reset psta 4
A compound with cell dimensions a=20.515, b=21.31,
c=17.128 is searched over 1/512 of the unit cell near the
origin, in steps of 1/128 of each cell edge. The resulting
RMAP is then searched for maxima. The D function calculated
is weighted by 0.1 in terms of 1/R and by 0.9 in terms of
C.





